
We do not diagnose disease or recommend a treatment protocol or dietary supplement for the treatment of disease. You should share this information with your physician who can determine what nutrition and disease treatment regimen is best for you. Ask your physician any questions you have concerning your medical condition.
You can search this site or the web for topics of interest that I may have written (use Dr Simone and topic). “We provide truthful information without emotion or influence from the medical establishment, pharmaceutical industry, national organizations, special interest groups or government agencies.” Charles B Simone, M.MS., M.D.
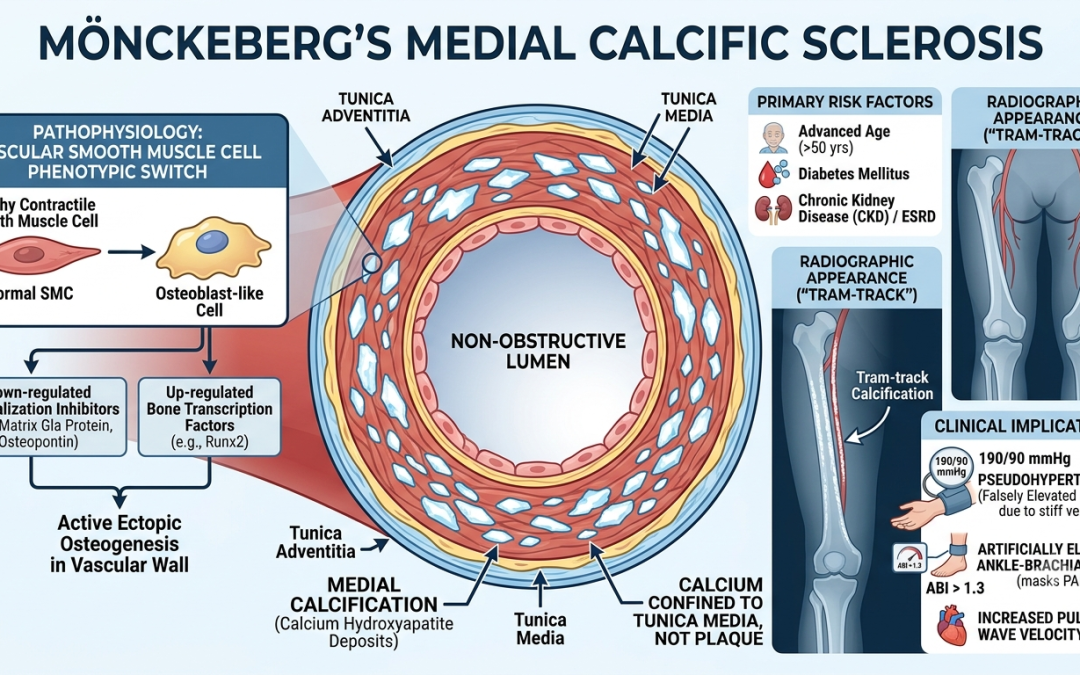
Mönckeberg’s arteriosclerosis (also known as medial calcific sclerosis) is a form of vessel hardening characterized by dystrophic calcification within the tunica media of medium- and small-sized muscular arteries.
Unlike atherosclerosis, Mönckeberg’s disease is traditionally considered non-obstructive because the calcium deposits are confined to the medial layer and do not typically encroach upon the vessel lumen or disrupt the endothelial lining. However, it significantly reduces arterial compliance, turning muscular vessels into rigid, non-distensible “pipestems.”
Etiology & Pathophysiology
The exact molecular pathways remain an area of ongoing study, but Mönckeberg’s is no longer viewed as a passive, inevitable consequence of aging. Instead, it is an active, regulated process resembling ectopic osteogenesis (bone formation) within the vascular wall.
-
Phenotypic Switch: Vascular smooth muscle cells (VSMCs) undergo a phenotypic transition into osteoblast-like cells. This switch is driven by down-regulated mineralization inhibitors (e.g., matrix Gla protein, osteopontin) and up-regulated bone-associated transcription factors (e.g., Runx2).
-
Primary Risk Factors:
-
Advanced Age: Most commonly observed in individuals older than 50.
-
Diabetes Mellitus: Autonomic neuropathy and chronic hyperglycemia heavily accelerate medial calcification.
-
Chronic Kidney Disease (CKD) / ESRD: Disrupted calcium-phosphate homeostasis, hyperphosphatemia, and secondary hyperparathyroidism are potent triggers.
-
Histological cross-section showcasing dense basophilic calcification within the tunica media layer.. Source: Wikipedia
Clinical Implications & Complications
While the disease itself does not cause direct ischemic occlusion, the loss of arterial elasticity induces severe downstream hemodynamic consequences:
-
Pseudohypertension & Diagnostic Distortion: The rigid arterial walls resist compression during blood pressure measurements, leading to falsely elevated systolic readings. More critically, it artificially elevates the Ankle-Brachial Index (ABI)—often resulting in readings greater than 1.3 or 1.4—which can mask underlying peripheral artery disease (PAD).
-
Increased Pulse Wave Velocity: Loss of medial compliance accelerates the systolic pressure wave, causing it to reflect back to the heart early. This increases cardiac afterload, leading to left ventricular hypertrophy (LVH) and isolated systolic hypertension.
-
Co-existence with Atherosclerosis: While distinct pathophysiologically, the systemic environments that foster Mönckeberg’s (CKD, diabetes) also aggressively promote intimal atherosclerosis. When combined, rigid walls make endovascular interventions significantly more challenging.
-
Calciphylaxis Link: In severe CKD populations, advanced medial calcification can overlap with calcific uremic arteriolopathy, leading to ischemic skin necrosis.
Diagnosis
Because it is usually asymptomatic on its own, Mönckeberg’s is frequently an incidental finding:
-
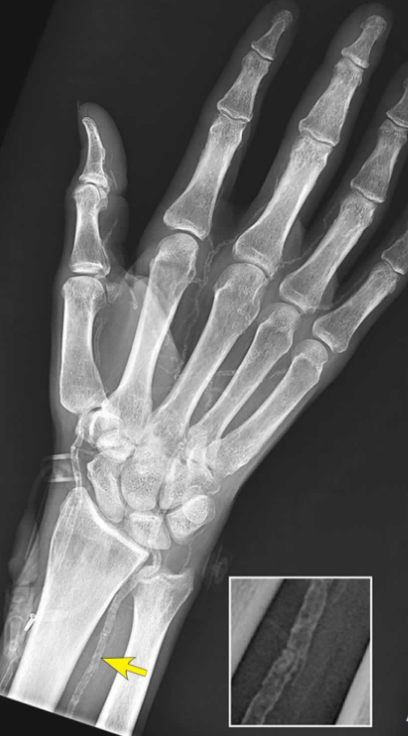
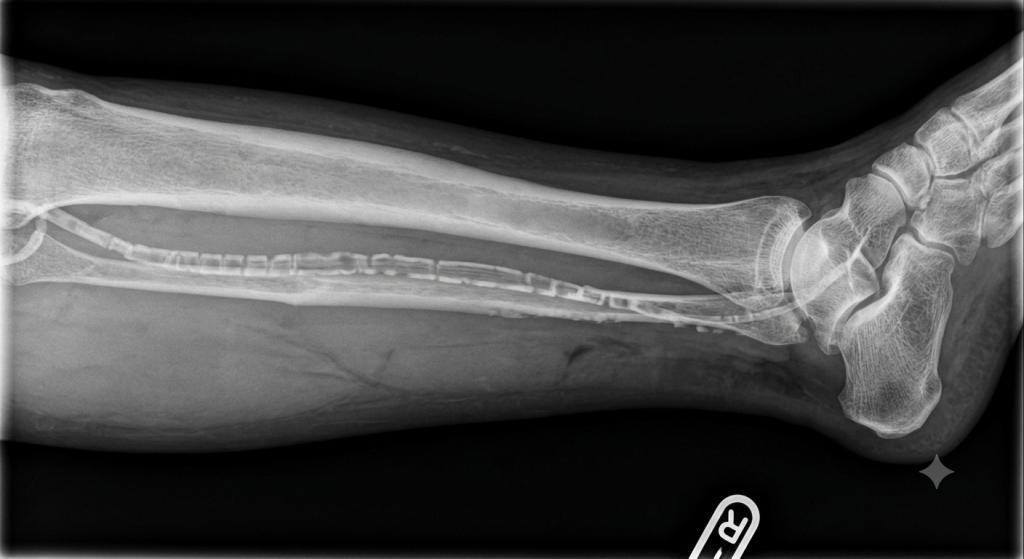
Plain Radiography: Appears as distinct, continuous, circumferential “tram-track” calcifications outlining the course of peripheral vessels (e.g., pelvic, femoral, or digital arteries), independent of any plaque formation.
-
-
Mönckeberg’s medial calcific sclerosis in the upper extremity. Source: Cleveland Clinic Journal of Medicine
-
-
Toe-Brachial Index (TBI): Because digital arteries are less susceptible to severe non-compressible calcification than larger ankle vessels, TBI is preferred over ABI when evaluating concurrent PAD in these patients.
Treatments & Management
There is no specific, targeted therapy to reverse existing medial calcification. Management centers entirely on addressing underlying metabolic drivers and mitigating cardiovascular risk:
-
Metabolic & Glycemic Control: Strict optimization of blood glucose levels to slow the progression of VSMC phenotypic shifting.
-
Mineral Optimization (CKD Patients): Aggressive management of phosphate and calcium balance using non-calcium-based phosphate binders (e.g., sevelamer), calcimimetics, and careful calibration of vitamin D analogues.
-
Surgical Nuances: When peripheral revascularization is required for comorbid PAD, the brittle, rigid nature of Mönckeberg-affected vessels elevates the risk of arterial dissection, poor stent expansion, and vessel recoil during angioplasty.
WHAT ARE THE NATURAL NON-CALCIUM BASED PHOSPHATE BINDERS, CALCIMIMETIC, AND WHAT SHOULD BE THE LEVEL OF SERUM VITAMIN D?
DOES SUPPLEMTAL VITAMIN D CAUSE MONCKEBERG’s DISEASE?
1. Natural Non-Calcium Based Phosphate Binders
In clinical medicine, prescription non-calcium binders like Sevelamer (an ion-exchange resin) and Lanthanum carbonate or Sucroferric oxyhydroxide (metal-based binders) are the standard of care. When looking strictly at naturalor over-the-counter options, choices are highly limited because binding phosphate requires a dense positive charge to trap negative phosphate ions (PO43−?).
-
Chitosan: This is a natural, indigestible polysaccharide derived from the exoskeletons of crustaceans (shrimp, crabs). Structurally similar to sevelamer, its positively charged amine groups naturally bind to negative ions. While historical interest surged around using chitosan gum to trap phosphate in saliva, large trials showed it had minimal clinical impact due to mass-balance limits (a few milligrams of chitosan cannot bind hundreds of milligrams of dietary phosphate). However, specialized high-dose chitosan supplements (used under strict clinical guidance) have shown a modest ability to bind intestinal phosphate as an alternative for patients completely intolerant to prescription binders.
-
Dietary Fiber Modifiers: High-fiber diets can slightly alter transit times and decrease the overall bioavailability of organic phosphorus, though they are not true “binders” in the biochemical sense.
2. Calcimimetics
Calcimimetics are a class of medications that target the Calcium-Sensing Receptors (CaSR) located on the chief cells of the parathyroid gland.
Instead of increasing serum calcium, these drugs physically bind to the CaSR and increase its sensitivity to existing extracellular calcium. The parathyroid gland is essentially “tricked” into thinking serum calcium levels are higher than they actually are.
-
Mechanism: It suppresses the synthesis and secretion of Parathyroid Hormone (PTH).
-
Clinical Impact: By lowering severe secondary hyperparathyroidism without introducing exogenous calcium, calcimimetics help prevent the catastrophic bone-to-mineral shifting that drives Mönckeberg medial sclerosis and calciphylaxis.
-
Common Examples: Cinacalcet (oral) and Etelcalcetide (intravenous, administered during hemodialysis).
3. Targeted Serum Vitamin D Levels
In the context of vascular health and chronic kidney disease-mineral and bone disorder (CKD-MBD), target levels depend entirely on which form of Vitamin D is being measured:
-
Nutritional Vitamin D [25(OH)D]: The general consensus for maintaining systemic bone health and preventing deficiency is to maintain serum levels between 30 ng/mL and 50 ng/mL. Levels falling below 20 ng/mL represent true deficiency, which triggers secondary hyperparathyroidism and bone resorption.
-
Active Vitamin D [ / Calcitriol]: Active vitamin D levels are not typically targeted by a standard number; rather, active vitamin D analogs (e.g., Calcitriol, Paricalcitol) are strictly dosed and titrated based on achieving target ranges for PTH, Serum Calcium, and Serum Phosphate to prevent a high calcium-phosphate product ().
4. Does Supplementing Vitamin D Cause Mönckeberg Disease?
Vitamin D exhibits a highly sensitive, biphasic (U-shaped) dose-response curve regarding vascular calcification.
Vascular Calcification Risk
| \ / <-- Hypervitaminosis D (Hypercalcemia + Hyperphosphatemia)
| \ /
| \_________/ <-- Optimal Window (25(OH)D: 30-50 ng/mL)
| Deficiency
+-------------------> Vitamin D Status
The High-Dose Risk (Hypervitaminosis)
Yes, in excessive, pharmacological doses, Vitamin D can directly induce Mönckeberg’s medial sclerosis. In fact, giving laboratory animals massive doses of active Vitamin D (Calcitriol) is the standard experimental model used to create Mönckeberg disease for research.
When active Vitamin D levels are excessively high, it over-stimulates intestinal absorption of both calcium and phosphate and drives osteoclastic bone resorption. This causes severe hypercalcemia and hyperphosphatemia. The excess minerals overwhelm systemic tissue inhibitors (like matrix Gla protein), forcing vascular smooth muscle cells (VSMCs) in the tunica media to transition into osteoblast-like cells and lay down calcium hydroxyapatite.
The Low-Dose Risk (Deficiency)
Paradoxically, Vitamin D deficiency also accelerates medial vascular calcification. When serum 25(OH)D drops too low, the parathyroid gland goes into overdrive (severe secondary hyperparathyroidism). This chronic elevation of PTH constantly leaches calcium and phosphorus out of the skeleton and into the bloodstream, raising the local mineral product within the vessel walls. Furthermore, healthy physiological levels of vitamin d receptor (VDR) activation inside the endothelium are protective; they suppress the inflammatory pathways that cause smooth muscle cells to osteogenically transform.
The Bottom Line on Supplementation
Standard, nutritional over-the-counter supplementation (e.g., 1,000 to 5,000 IU of Cholecalciferol D3? daily) taken to maintain a healthy blood level of 30–50 ng/mL does not cause Mönckeberg disease in individuals with normal renal function. The danger arises under two specific conditions:
-
Massive Mega-Dosing: Taking extreme, unregulated quantities that induce hypercalcemia.
-
Advanced Renal/CKD Environment: In patients with renal failure, the body’s ability to excrete phosphate is broken. Even standard therapeutic doses of active vitamin D sterols must be carefully managed with calcimimetics and non-calcium binders, because any slight elevation in calcium or phosphate can instantly precipitate out into the non-compliant, rigid medial layers of the arterial tree.
Can Mönckeberg’s disease be reversed if the patient stops taking excess Vitamin D
Clinically, established Mönckeberg’s disease is widely considered irreversible in human patients, even after stopping the excess Vitamin D.
Once vascular smooth muscle cells (VSMCs) have fully transitioned into osteoblast-like cells and laid down an organized matrix of hydroxyapatite (calcium phosphate) within the tunica media, the structural “pipestem” change is permanent. Human tissue lacks a reliable, natural metabolic pathway to safely demineralize or “melt away” crystallized calcium from the elastic layers of a blood vessel without destroying the vessel itself.
However, stopping the excess Vitamin D alters the disease trajectory significantly:
What Happens When Excess Vitamin D is Stopped?
1. Arrest of Disease Progression
While existing calcification will not disappear, removing the source of hypervitaminosis D drops serum calcium and phosphate levels back into physiological ranges. This cuts off the raw material supply driving the mineralization process and halts the phenotypic signaling causing remaining healthy VSMCs to transform into bone-producing cells. Progression stops.
2. Time-Dependent Soft Tissue Clearances
While the vessels remain calcified, other soft tissues can see real recovery. If the hypervitaminosis D induced acute hypercalcemia, calcium deposits in the kidneys (nephrocalcinosis), stomach lining, or lungs may slowly mobilize and clear once mineral homeostasis is restored, preserving long-term organ function.
3. Animal Models vs. Human Realities
In laboratory animal models (like rats) where Mönckeberg’s is rapidly induced via a massive, short-term burst of toxic active Vitamin D, scientists have observed mild regression of medial calcification over time once the toxin is removed. However, this is largely attributed to the highly efficient macrophages and rapid bone/tissue remodeling capabilities specific to young rodents. In humans, who typically accumulate this damage over months or years, this natural regressive capacity does not translate.
Are There Any Therapies That Can Reverse It?
Currently, there are no FDA-approved or clinically proven therapies to reverse Mönckeberg’s or systemic medial vascular calcification. Standard treatments focus strictly on stabilizing the vessels and protecting the heart from the resulting stiffness.
Experimental and Emerging Research
A few therapeutic pathways are being actively researched to see if reversal can ever be safely achieved:
-
Chelation Therapy (e.g., EDTA): In vitro and animal studies show that powerful chelating agents can physically extract calcium from hydroxyapatite and calcified aorta tissues. However, systemically using EDTA in human clinical trials has yielded highly controversial and inconsistent results. A major risk is that systemic chelation cannot distinguish between the “bad” calcium in an artery and the “good” calcium stabilizing the skeleton, often leading to bone loss or severe electrolyte imbalances.
-
Pyrophosphate and Matrix Gla Protein (MGP) Up-regulation: Researchers are looking into ways to medically boost natural calcification inhibitors. Sodium thiosulfate (sometimes used in calciphylaxis) and high-dose Vitamin K2 (which activates MGP to direct calcium away from vessels and into bones) are being studied to see if they can prevent progression, though they cannot actively dissolve heavy, pre-existing structural medial sheets.
How much K2?
When considering Vitamin K2 (specifically Menaquinone-7 or MK-7, which has a longer half-life and better vascular bioavailability than MK-4), the optimal dosage depends heavily on whether you are looking at general health maintenance or the aggressive doses used in vascular clinical trials.
The Council for Responsible Nutrition (CRN) establishes the upper safety limit for adults at 375 mcg daily.
1. The Vascular Clinical Trial Doses
To specifically target the activation of Matrix Gla Protein (MGP)—the primary protein responsible for keeping calcium out of arterial walls—clinical trials use much higher doses than a standard daily multivitamin.
-
The Cardiovascular Standard: 360 mcg daily
The rigorous VitaK-CAC randomized controlled trial evaluated patients with established coronary artery calcification. Participants were given 360 mcg of MK-7 daily for 24 months.
-
The Result: The high-dose K2 group experienced nearly one-third less progression of arterial calcification and a 42% reduction in calcium mass accumulation compared to the placebo group.
-
-
The Chronic Kidney Disease (CKD) Standard: 90 to 360 mcg daily
For patients struggling with mineral metabolic shifts (who are at the highest risk for Mönckeberg disease), studies typically titrate between 90 mcg and 360 mcg daily, often paired with low-dose nutritional Vitamin D3 to safely maintain bone density without spilling mineral product into the vasculature.
2. General Physiological Maintenance Doses
If the goal is simply standard nutritional support to ensure baseline activation of bone and vascular proteins, the dosages are lower:
-
Standard Therapeutic Range: 90 mcg to 180 mcg daily of MK-7. This range is widely accessible over-the-counter and matches historical epidemiological data (like the Rotterdam Study) showing reduced aortic calcification risks in populations with high dietary K2 intake.
Critical Safety Warning: The Warfarin Exception
Absolute Contraindication: If a patient is taking the blood thinner Warfarin (Coumadin), they cannot take high-dose Vitamin K2 supplements without severe medical risks.
Warfarin works by intentionally blocking the Vitamin K cycle to thin the blood. Introducing exogenous Vitamin K2 (especially at doses like 360 mcg) will directly neutralize Warfarin’s mechanism of action, thickening the blood and potentially causing a life-threatening blood clot or stroke.
(Note: Newer classes of blood thinners, such as DOACs/NOACs like Eliquis or Xarelto, do not rely on Vitamin K inhibition, but any supplementation strategy should still be managed directly under a physician’s oversight).
If a patient is taking 325 mg aspirin, 4 grams of omega 3, and 1,000 IU of Vitamin E daily for blood thinning, should vitamin K be taken as a supplement?
Yes, a patient taking 325 mg Aspirin, 4 grams of Omega-3, and 1,000 IU of Vitamin E can generally take a Vitamin K2 supplement, but it requires careful management.
Unlike Warfarin (Coumadin), none of these three agents rely on blocking the Vitamin K pathway to thin the blood. Therefore, Vitamin K2 will not neutralize their therapeutic effects.
However, introducing Vitamin K2 into this specific regimen creates a delicate balancing act between antiplatelet activity, systemic bleeding risks, and vascular calcification protection.
1. Interaction Profile: How They Behave Together
To understand how to safely introduce Vitamin K2, it helps to look at exactly how the patient’s current regimen is working:
Agent Mechanism Interaction with Vitamin K2 Aspirin (325 mg) Antiplatelet: Irreversibly inhibits the COX-1 enzyme, stopping platelets from sticking together to form a clot. This is a full adult treatment dose (not a baby aspirin). None. Vitamin K manages clotting factors (proteins in the blood), not blood platelets. Omega-3 (4 grams) Antiplatelet / High Dose: At 4 grams daily (prescription-grade dosing), it reduces platelet aggregation and lowers fibrinogen. None. It operates via fatty acid membrane displacement, completely independent of Vitamin K. Vitamin E (1,000 IU) Anticoagulant & Antiplatelet: This is the critical variable. At doses above 800 IU, Vitamin E acts as a mild Vitamin K antagonist. It can interfere with how the liver uses Vitamin K, naturally extending bleeding times. Direct Metabolic Interaction. High-dose Vitamin E actively opposes Vitamin K. 2. The Clinical Trade-Offs: Benefits vs. Bleeding Risks
When evaluating whether to add Vitamin K2 to this specific stack, there are two distinct physiological forces at play:
The Risk: High Bleeding Profile
The patient is already on an incredibly potent, multi-pathway “natural” and pharmaceutical thinning regimen. The combination of full-dose Aspirin, high-dose Omega-3s, and mega-dose Vitamin E creates a significantly elevated risk for bruising, nosebleeds, or internal GI bleeding.
The Benefit: Restoring the Vitamin E-K Balance
Because 1,000 IU of Vitamin E is an extreme dose that induces a localized Vitamin K deficiency in the liver, adding a Vitamin K2 (MK-7) supplement can actually help normalize systemic mineral distribution.
Vitamin K2 will activate Matrix Gla Protein (MGP) to protect the patient’s arteries from the Mönckeberg-style calcification discussed earlier, without completely overriding the antiplatelet protection provided by the Aspirin and Omega-3s.
3. How to Approach Supplementation Safely
If a physician decides that protecting the patient’s arteries from medial calcification outweighs or balances the bleeding risks, the strategy should be highly calculated:
-
Avoid Vitamin K1: Do not take high doses of Vitamin K1 (phytonadione), as it goes straight to the liver and focuses heavily on blood coagulation.
-
Target Vitamin K2 (MK-7): Choose the MK-7 form of Vitamin K2. It bypasses primary liver coagulation centers and distributes preferentially to the blood vessels and bones, maximizing vascular protection while minimizing changes to baseline bleeding times.
-
Consider Lowering Vitamin E: From a metabolic standpoint, reducing Vitamin E from the mega-dose of 1,000 IU down to a standard structural dose (e.g., 200–400 IU) would naturally reduce the localized Vitamin K inhibition, making a K2 supplement far more effective and drastically lowering the patient’s overall systemic bleeding risk.
-
Mandatory Lab Tracking: Before and after starting Vitamin K2, a physician should run a Prothrombin Time (PT/INR) test and a complete blood count (CBC) to monitor exactly how the patient’s clotting factors and platelets are responding to the combination.
-

